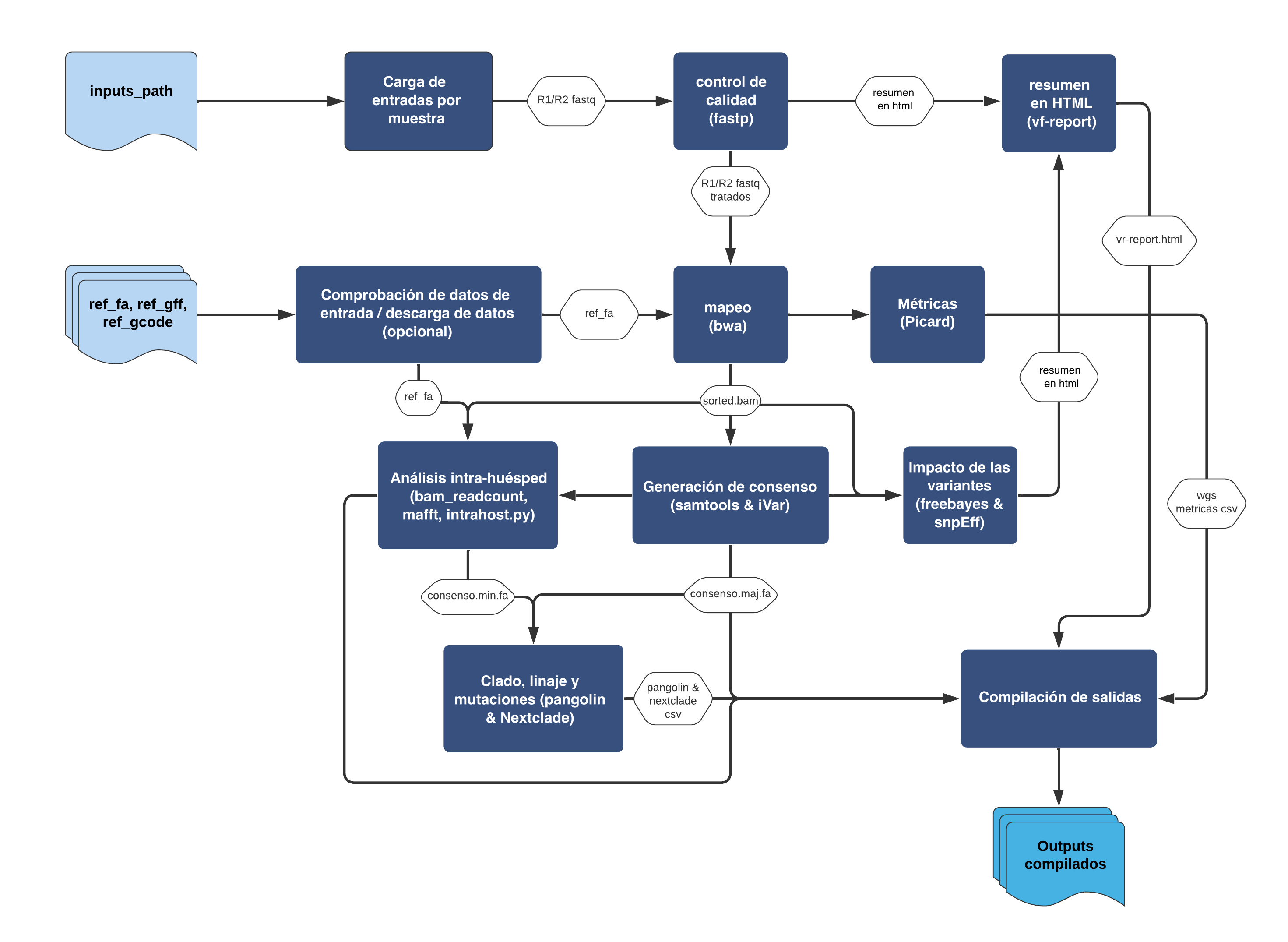
De que se trata
ViralFlow es un workflow elaborado para profesionales de la salud, con el objetivo de ejecutar todas las etapas de un análisis genómico viral por referencia.
El código fue escrito en el lenguaje de workflow Nextflow, y puede ser aplicado para diferentes virus, donde el usuario, después de instalar la herramienta, necesita rodar solamente una línea de código.
El grupo que desarrolló el ViralFlow no ofrece soporte, por el momento, para errores relacionados con la aplicación del código en otras plataformas. Hasta el presente, el código ha sido utilizado solamente en datos generados por secuenciadores Illumina, empleando estrategia de reads por pares (paired-end) o no emparejados (single-end).
La publicación de la primera versión del ViralFlow puede ser verificada en el link a continuación.. La publicación de la vérsion 1.0, que incluye las funciones descritas en este documento, puede ser verificada en el link a continuación..
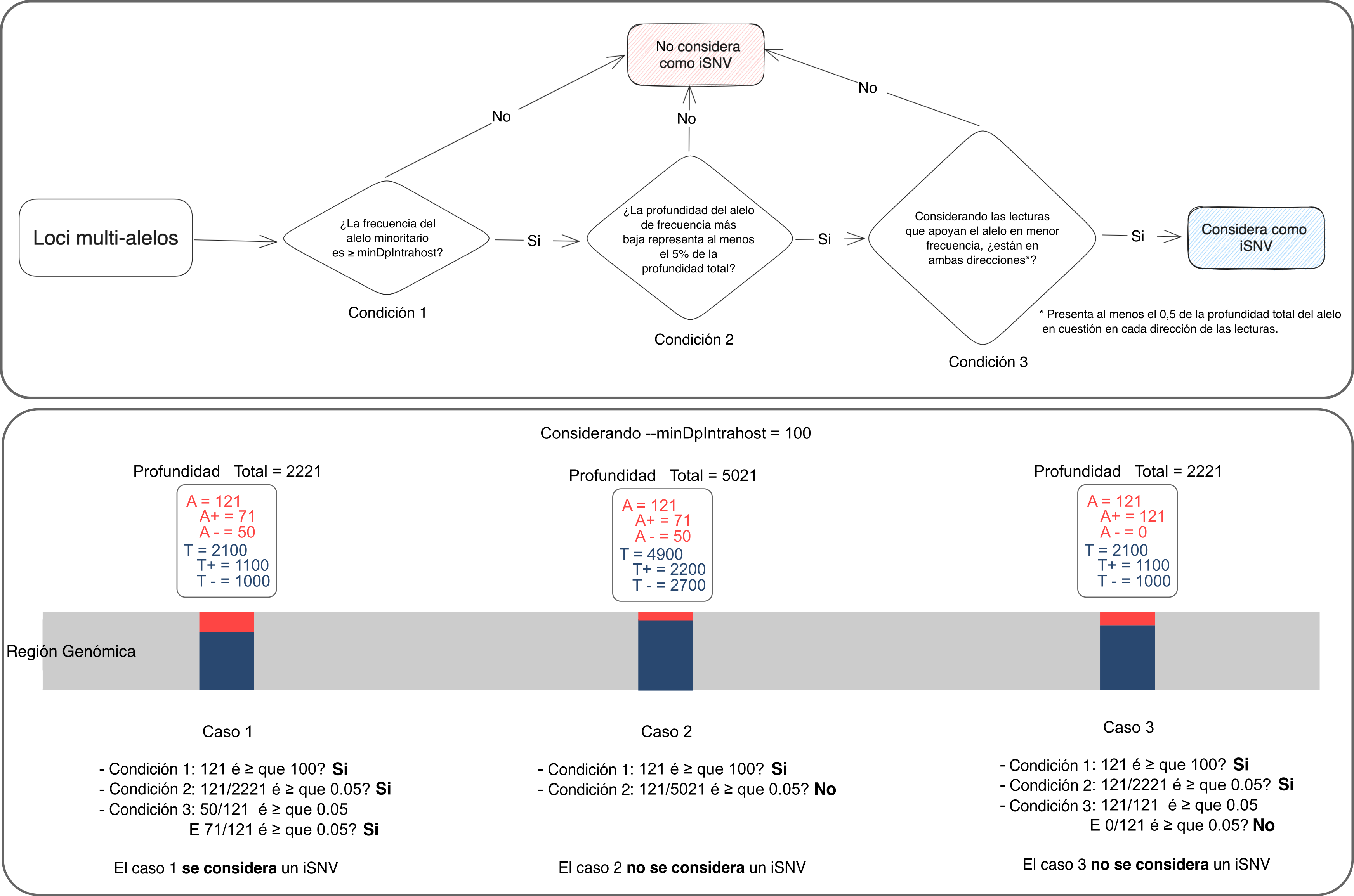
Análisis intra-huésped
El ViralFlow presenta un algoritmo propio para detectar regiones de iSNV (intrahost Single Nucleotide Variant).
Para que un determinado locus multi alélico tenga un alelo con menor frecuencia, considerado iSNV, tres condiciones deben cumplirse, estas condiciones están esquematizadas en la parte superior de la figura que se muestra a continuación.
Siguiendo esta lógica, la pare inferior de la figura a continuación representa 3 sitios multi alélicos y ejemplifica la lógica para considerar cuáles de ellos serian considerados iSNVs.
dependencias
Para la instalación y ejecución del ViralFlow, será necesario disponer de al menos 4,4 Gb de memoria, para la elaboración de los contenedores, e de tres dependencias:
- pip
- Git
- uidmap
La lista con las versiones de cada herramienta utilizada en el ViralFlow puede ser verificada en el siguiente link.. Adicionalmente, para la instalación con un ambiente controlado, se usa el Conda. El ViralFlow utiliza la herramienta de creación de contenedores Singularity y del lenguaje de workflow Nextflow. Para garantizar el funcionamiento correcto del ViralFlow es esencial tener las versiones adecuadas del Singularity y del Nextflow, y ambos pueden ser instalados simultáneamente con el ViralFlow.
-
01 Conda
Conda es un administrador de paquetes de código abierto. Fue inicialmente desarrollado para gerenciar ambientes de paquetes de lenguaje Python, permitiendo el control de versiones.
Para que todos los usuarios utilicen el ViralFlow con las versiones exactas del Singularity y del Nextflow, hemos creado un ambiente con estas versiones, permitiendo una estandarización en los ambientes de instalación del ViralFlow, así como la resolución de eventuales errores.
-
02 Singularity v3.11.4
Singularity es una herramienta que ejecuta la virtualización a nivel del sistema operacional, tarea popularmente conocida como creación de contenedores.
Para un pipeline computacional funcionar, como el ViralFlow, se necesita de varias dependencias (otras herramientas y bibliotecas del sistema operacional); y para evitar que el usuario necesite instalar todas las dependencias manualmente en su máquina, utilizamos el sistema de contenedores.
Con la creación de contenedores, el usuario solo necesita instalar una herramienta, en este caso el Singularity, y después construir los contenedores basándose en archivos de receta (en el caso del ViralFlow, también fue automatizado con el script setupContainers.sh). De esta forma, diferentes grupos de investigación pueden rodar la herramienta en el mismo ambiente computacional, sin cambios en el comportamiento del ViralFlow, garantizando la reproductibilidad de los resultados. -
03 Nextflow 22.04
Nextflow es un administrador de workflows de bioinformática que permite el desarrollo de workflows portátiles y reproducibles.
Nextflow permite la ejecución de los workflows en diversos ambientes computacionales, como ambientes locales, de computación de alta performance (HPC), servicios de computación en la nube, AWS y Google Cloud, y también, en sistemas de orquestación de contenedores como Kubernetes.
Adicionalmente, el Nextflow proporciona soporte para gerenciar las dependencias de Conda, Docker, Podman o Singularity.
¿Cómo instalar (MacOS)?
Considerando las limitaciones del uso del Singularity en el MacOS, para rodar el ViralFlow en este tipo de sistema se sugiere utilizar un software de virtualización Linux llamado Lima. De esta forma, el usuario debe seguir tres pasos para la instalación del Lima, y después continuar con la guía de instalación del Ubuntu
Instalando el Lima
Instale el Lima utilizando el administrador de paquetes Homebrew:
brew install lima
Instalando la instancia Ubuntu
Siga el paso a paso de la instalación de la instancia Ubuntu introduciendo el comando a seguir
limactl start
Inicie el Ubuntu
Cuando la maquina virtual sea instalada, utilice el comando a continuación para iniciar el Ubuntu y siga los pasos de instalación del ViralFlow normalmente.
lima
¿Cómo instalar (Ubuntu)?
Para realizar la instalación del ViralFlow, deben seguirse cuatro pasos: Instalar las dependencias del sistema, en el caso de haberlo hecho antes; Instalar el Conda; Instalar el ViralFlow y, finalmente, montar los contenedores para los análisis. Este proceso es realizado una única vez.
La instalación y uso del ViralFlow ya fue probada con los sistemas:
- Ubuntu 20.04 LTS;
- Ubuntu 22.04 LTS;
Instalando as dependencias del sistema
En el caso de no tener el instalador de dependencias pip, el sistema de control de versiones git y el paquete uidmap, usted debe realizar la instalación de las siguientes líneas de código:
sudo apt update -y && \
sudo apt upgrade -y && \
sudo apt install curl git python3-pip uidmap -y
Instalando y configurando el ambiente micromamba
Recomendamos el administrador de ambientes micromamba por su paralelización al realizar el download e instalación de las dependencias. En el caso de que usted utilice otro administrador de ambientes, como conda, miniconda, mamba, etc, puede continuar con la instalación del ViralFlow ignorando esta etapa, sin embargo, pueden ocurrir conflictos durante la instalación por las versiones de las dependencias especificadas, y sus respectivas disponibilidades en diferentes canales de recursos.
En el caso de no tener el administrador de ambientes micromamba instalado, podrá hacerlo y configurar con las siguientes líneas de código:
cd $HOME
curl -Ls https://micro.mamba.pm/api/micromamba/linux-64/1.5.7 | tar -xvj bin/micromamba
./bin/micromamba shell init -s bash -p ~/micromamba
source ~/.bashrc
micromamba activate
Instalando el ViralFlow
Si usted ya dispone de las dependencias citadas y el conda instalado, podrá instalar el ViralFlow con 5 líneas de código:
git clone https://github.com/WallauBioinfo/ViralFlow
cd ViralFlow/
micromamba env create -f envs/env.yml
micromamba activate viralflow
pip install -e .
Construyendo los contenedores
Todas las etapas del ViralFlow son ejecutadas en ambientes controlados, donde cada herramienta tendrá la misma versión independientemente del grupo de investigación que haga uso de ella. El ViralFlow posee un método propio para realizar toda esa construcción de ambientes, rodando apenas una línea de código. Para que la construcción de los contenedores ocurra, el ViralFlow necesita que la herramienta “unsquashfs” se encuentre disponible en el directorio “/usr/local/bin/”. Durante el proceso de construcción de los contenedores, se realiza una etapa de verificación de la presencia de unsquashfs. Si el sistema del usuario no presenta el unsquashfs en el lugar ideal, el ViralFlow irá sugerir los pasos necesarios para la solución de este problema. Para evitar este problema, el usuario puede crear un link simbólico de la herramienta “unsquashfs” para el directorio “/usr/local/bin/”, y para ello, el siguiente comando debe ejecutarse:
sudo ln -s /usr/bin/unsquashfs /usr/local/bin/unsquashfs
Después de garantizar que el unsquashfs se encuentra en el lugar adecuado, ejecuta el comando para la construcción de los contenedores:
viralflow -build_containers
Como ejecutar (Guía rápido)
El ViralFlow dispone de dos modos de uso: sars-cov2 e custom. Independientemente del modo de uso, el usuario debe realizar los caminos absolutos (todo el camino hasta el directorio o archivo que será indicado para el pipeline ex. /home/usuario/teste/) para cada archivo de entrada, en el caso de no hacerse, el pipeline será interrumpido durante la ejecución. La explicación de cada argumento puede ser verificada aquí..
La herramienta snpEff presente en el ViralFlow ya viene configurada solamente con el genoma NC_045512.2 del virus SARS-CoV-2. En el caso de querer el análisis del snpEff para otros virus, deberá actualizarse el banco del snpEff con la siguiente línea de comando; ejemplo Dengue:
viralflow -add_entry_to_snpeff --org_name Dengue --genome_code NC_001474.2
sars-cov2
En este modelo, el análisis es realizado considerando el genoma de referencia NC_045512.2, y posee como análisis adicionales la inclusión de linajes con la herramienta Pangolin, y la inclusión de clados y mutaciones con la herramienta Nextclade. Para ello, el usuario necesita construir un archivo con los parámetros del análisis, como el ejemplo a continuación.
viralflow -run --params_file test_files/sars-cov-2.params
custom
En este modelo, el análisis es realizado considerando los archivos para el virus que el usuario desea analizar. De esta forma, el usuario es responsable por pasar cada uno de los archivos necesarios para el análisis. En el caso de que el usuario desee realizar el análisis del snpEff, debe pasar el código del refseq del genoma viral para el análisis:
viralflow -run --params_file test_files/denv.params
Actualización pangolín
Periódicamente, la herramienta Pangolin actualiza el banco de linajes, así como la filogenia de la clasificación de Usher, las constelaciones de mutaciones de scorpio y el modelo utilizado del pangoLearn. Para realizar la actualización de la herramienta y/o de sus bases de datos, basta ejecutar el ViralFlow con alguno de los comandos:
#actualiza la herramienta y las bases de datos
viralflow -update_pangolin
#solo actualiza las bases de datos
viralflow -update_pangolin_data
Argumentos
| Argumento | Valor estandard (default) | Descripción |
|---|---|---|
| virus | sars-cov2 | Tipo de análisis (sars-cov2 o custom) |
| primersBED | null | Camino absoluto del archivo BED con informaciones de los primers usados en la amplificación genómica (opcional). |
| outDir | launchDir/output/ | Camino absoluto para el directorio donde serán almacenados los resultados. |
| inDir | launchDir/input/ | Camino absoluto para el directorio con los datos de entrada (directorio con los archivos FASTQ). |
| runSnpEff | true | Necesidad de ejecutar la herramienta snpEff (true or false). |
| writeMappedReads | true | Necesidad de generar los archivos FASTQ con las lecturas de secuenciación que mapearon en el genoma de referencia. |
| minLen | 75 | Tamaño mínimo que el read debe tener para no ser eliminado por el FastP. |
| depth | 5 | Profundidad mínima que una región debe ser secuenciada para ser considerada en el consenso. |
| mapping_quality | 30 | Umbral de calidad de mapeo para llamada de variantes. |
| base_quality | 30 | Umbral de calidad de base para llamada de variantes. |
| minDpIntrahost | 100 | Profundidad mínima que una región debe tener para ser considerada en el análisis de intrahuésped. |
| trimLen | 0 | Número de bases que deben ser cortadas (“trimadas”) en las dos extremidades de los reads. |
| refGenomeCode | null | Código refseq del genoma a ser utilizado em el análisis custom. |
| referenceGFF | null | Arquivo GFF do genoma a ser utilizado na análise custom. |
| referenceGenome | null | Archivo fasta del genoma a ser utilizado em el análisis custom. |
| nextflowSimCalls | null | Número de llamadas simultáneas que el nextflow puede realizar. |
| fastp_threads | 1 | Número de threads a ser utilizado em el análisis de la herramienta fastp. |
| bwa_threads | 1 | Número de threads a ser utilizado em el análisis de la herramienta bwa. |
| dedup | false | Este argumento habilita el modo de deduplicación del fastp. para activarlo, altere el valor para true en el archivo de prueba de parámetros. |
| ndedup | 3 | Cuando el modo de desduplicación está activo, se podrá usar niveles de precisión (1 - 6). Usted podrá alterar ese valor, pero recomendamos el estandard. Mientras más alto, más RAM y tiempo son consumidos. Para activarlo altere el valor de 1 para 6 en el archivo de prueba de parámetros. |
Archivos de Outputs
La herramienta ViralFlow produce dos tipos de outputs: especifico y compilado.
Especifico
Para cada muestra en el análisis será creado un directorio de resultados con el patrón “prefijo_results” donde el prefijo es un código creado a partir del nombre del archivo fastq de la muestra. Cada directorio cuenta con los siguientes resultados:
| Archivo | Descripción |
|---|---|
| prefixo.all.fa.pango.out.csv | Archivo tabulado con los resultados de la herramienta pangolin. |
| prefixo.ann.vcf | Archivo vcf en formato tsv (Tab separated value - valores separados por coma) con la anotación de las variantes hecha por la herramienta snpEff. |
| prefixo.depth5.all.fa.nextclade.csv | Archivo tabular con los resultados de la herramienta nextclade. |
| prefixo.depth5.amb.fa | Genoma consenso con nucleótidos ambiguos en posiciones de alelos múltiples. |
| prefixo.depth5.fa | Genoma consenso con nucleótidos mayoritarios. Consenso de la mayoría, normalmente depositado en el GISAID. |
| prefixo.depth5.fa.algn | Genoma consenso alineado con el genoma de referencia (siguiendo el mismo tamaño, mafft --keeplenght). |
| prefixo.depth5.fa.algn.minor.fa | Genoma consenso con nucleótidos minoritarios. |
| prefixo.depth5.fa.bc.intrahost.short.tsv | Archivo tabular resumiendo las posiciones genómicas donde las variantes intrahuésped aparecen. |
| prefixo.depth5.fa.bc.intrahost.tsv | Archivo tabular con todas las informaciones de las posiciones de variantes intrahuésped. |
| prefixo.mapped.R1.fq.gz | Archivo FASTQ R1 con los reads mapeados. |
| prefixo.mapped.R2.fq.gz | Archivo FASTQ R2 con los reads mapeados. |
| prefixo.metrics.genome.tsv | Archivo tabular con métricas de profundidad y cobertura del mapeamento. |
| prefixo.fastp.html | Archivo html con el resumen de los resultados de la herramienta fastp. |
| prefixo_snpEff_summary.html | Archivo html con el resumen de los resultados de la herramienta snpEff. |
| prefixo.sorted.bam | Archivo con el mapeamento sorteado de los reads de la muestra contra el genoma de referencia. |
| prefixo.tsv | Archivo vcf-like generado por el iVar. |
| prefixo.unmapped.R1.bam.fq | Archivo FASTQ R1 con los reads no mapeados. |
| prefixo.unmapped.R2.bam.fq | Archivo FASTQ R2 con los reads no mapeados. |
| prefixo.vcf | Archivo vcf. |
| metrics.alignment_summary_metrics | Archivo de texto con un resumen de diversas métricas del mapeamento. |
| nextclade.errors.csv | Archivo informando los errores del nextclade. |
| nextclade_gene_*.translation.fasta | Archivo fasta con las proteínas de cada gen. |
| snpEff_genes.txt | Archivo tabular con el número de variantes por impacto por gen. |
| wgs | Archivo textual con métricas del mapeamento, incluyendo profundidad por región. |
| prefixo_coveragePlot.png | Archivo PNG con la visualización gráfica de la cobertura del genoma. |
| prefixo_coveragePlot.svg | Archivo SVG con la visualización gráfica de la cobertura del genoma. |
| prefixo_coveragePlot.html | Archivo HTML con la visualización gráfica de la cobertura del genoma. |
| prefixo_snpPlot.png | Archivo PNG con la visualización gráfica de los SNPs detectados en la muestra con relación al genoma referencia. |
| prefixo_snpPlot.svg | Archivo SVG con la visualización gráfica de los SNPs detectados en la muestra con relación al genoma referencia. |
Compilado
Al final del análisis, es generado un directorio llamado COMPILED_OUTPUT con los resultados compilados, donde:
| Archivo | Descripción |
|---|---|
| errors_detected.csv | Muestras que con las que no fue posible generar consenso. |
| lineage_summary.csv | Es generado en el análisis del sars-cov-2, presenta el contaje de los linajes identificados en el análisis, incluyendo los linajes de genomas minoritarios. |
| major_summary.csv | Archivo tabular con un resumen de la profundidad, cobertura y linaje (en el caso de análisis de sars-cov-2) de los consensos mayoritarios. |
| minor_summary.csv | Archivo tabular con un resumen de la profundidad, cobertura y linaje (en el caso de análisis de sars-cov-2) de los consensos minoritarios. |
| mutations.csv | Archivo tabular con las mutaciones encontradas. |
| nextclade.csv | Archivo tabular con el resultado del nextclade (en el caso de análisis de sars-cov-2). |
| pango.csv | Archivo tabular con el resultado del pangolin (en el caso de análisis de sars-cov-2). |
| reads_count.csv | Archivo tabular con el contaje de reads por muestra. |
| seqbatch.fa | Archivo fasta con los consensos mayoritarios. |
| short_summary.csv | Archivo tabular con un resumen de la profundidad, cobertura y linaje (en el caso de análisis de sars-cov-2) de ambos consensos, con las siguientes columnas:
|
| wgs.csv | Archivo tabular con el resultado del picard. |
| VF_REPORT/index.html | Archivo html con la compilación de los resultados del fastp y del snpEff. |
Preguntas frecuentes (FAQ)
¿Debo montar los contenedores para cada análisis?
No, los contenedores solo deben ser reconstruidos en el caso de que ocurra alguna actualización de la herramienta.
¿El ViralFlow mostró un error debido a la falta de “unsquashfs” en el directorio “usr/local/bin/” que debo hacer?
Se debe crear un link simbólico para el “unsquashfs”, para ello ejecute la siguiente línea de comando: sudo ln -s/usr/bin/unsquashfs/usr/local/bin/unsquashfs.
¿Debo providenciar un archivo fasta de adaptadores?
Usted debe providenciar un archivo fasta con los primers de PCR solamente en análisis que utilizarán PCR para amplificación del material genético viral antes de la secuenciación.
Mi análisis de VialFlow reportó un error en la etapa de actualización del Pangolin, ¿por qué?
Esto puede ocurrir si el equipo de Pangolin lanza una versión con nuevas dependencias, en cuyo caso debe reconstruir el contenedor de Pangolin con la línea: singularity pull docker://staphb/pangolin
Mi instalación salió mal en la etapa de montaje de los contenedores, ¿por qué?
Lo que puede haber sucedido es que tengas una versión de singularity distinta a v3.11.4, puedes comprobarlo con singularity --version. Otro factor que puede causar errores es si su red de Internet bloquea los dominios de consulta para crear contenedores.
Instalé la herramienta, mis archivos están todos organizados siguiendo el manual, pero las muestras no son encontradas, ¿por qué?
El ViralFlow reconoce las muestras con los sufijos _R1 o _R2 de .fq.gz o .fastq.gz, verifique que sus muestras siguen el patrón necesario. Ejemplos:
- ART_R1.fq.gz, ART1_R2.fq.gz serán reconocidos como la muestra ART1;
- ART1_denv_R1.fastq.gz, ART1_denv_R2.fastq.gz serán reconocidos como la muestra ART1_denv.
¿Puedo ejecutar el ViralFlow en Windows?
A pesar de que algunas versiones de Windows integran una interface Linux por el WSL (Windows Subsystem for Linux), nuestro equipo de desarrollo no usó el ViralFlow en estos ambientes, por lo tanto, el usuario es libre para probar el workflow en este tipo de ambiente, pero no podrá contar con el soporte de nuestro equipo en el caso de errores provenientes de estas pruebas.
¿Cómo citar?
Actualmente hay dos publicaciones de ViralFlow, la primera, de 2022, que incluye las versiones “pre” 1.0, y la segunda, de 2024, que incluye la versión 1.0.
Dezordi, F.Z.; Neto, A.M.d.S.; Campos, T.d.L.; Jeronimo, P.M.C.; Aksenen, C.F.; Almeida, S.P.; Wallau, G.L.; on behalf of the Fiocruz COVID-19 Genomic Surveillance Network. ViralFlow: A Versatile Automated Workflow for SARS-CoV-2 Genome Assembly, Lineage Assignment, Mutations and Intrahost Variant Detection. Viruses 2022, 14, 217. https://doi.org/10.3390/v14020217
Silva, A. F., Silva Neto, A. M., Aksenen, C. F., Jeronimo, P. M. C., Dezordi, F. Z., Almeida, S. P., Costa, H. M. P., Salvato, R. S., Campos, T. L., Wallau, G. L., on behalf of the Fiocruz Genomic Network. (2024). ViralFlow v1.0—a computational workflow for streamlining viral genomic surveillance. NAR Genomics and Bioinformatics, 6(2), lqae056. https://doi.org/10.1093/nargab/lqae056