About
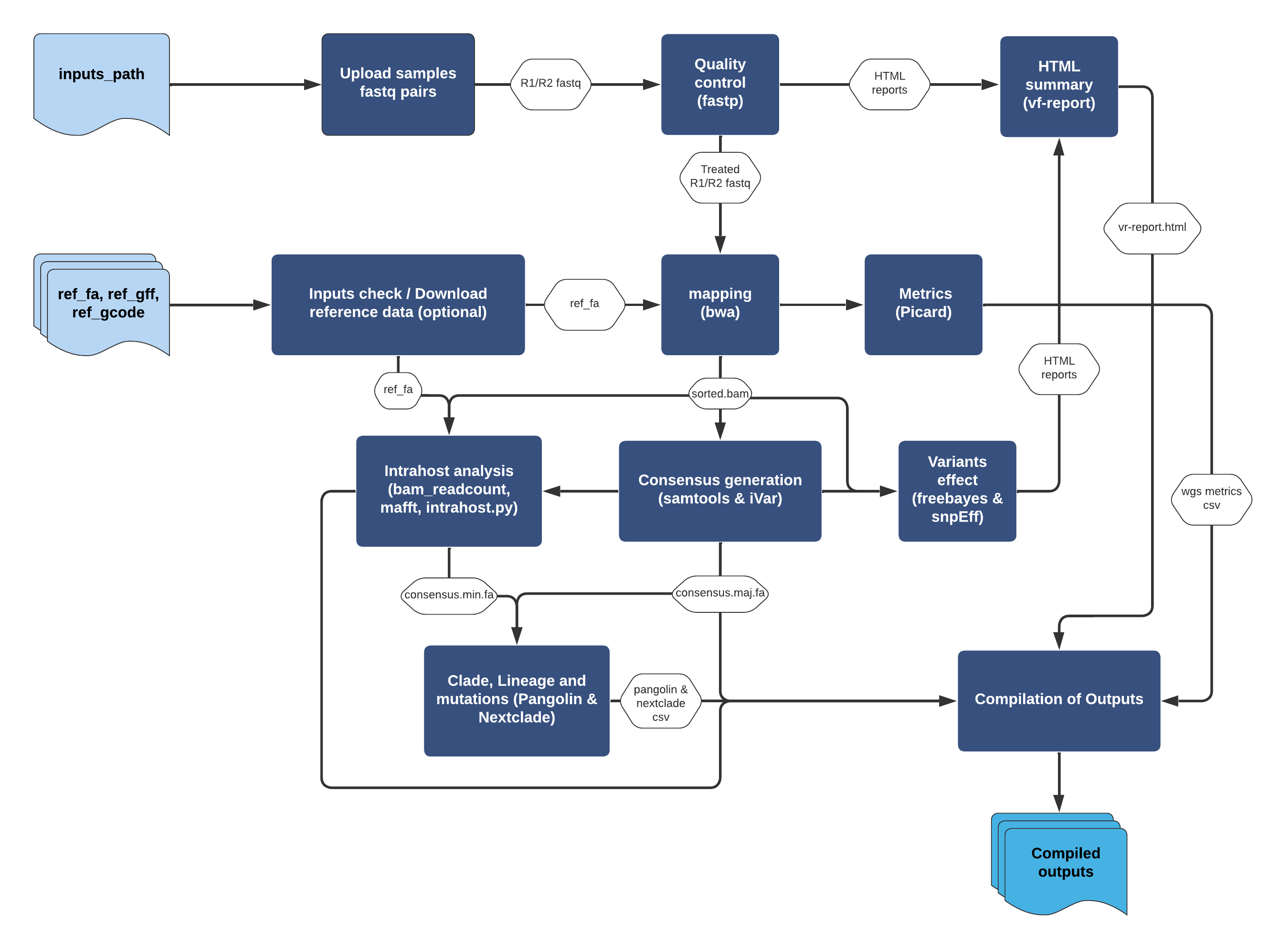
ViralFlow is a workflow developed for viral genomic surveillance that performs several genomic analyses based on reference genome assembly.
The current version of the pipeline was written in the Nextflow workflow language, and can be applied to different viruses. Only a single 1 command line is needed to run the entire workflow. To make using ViralFlow more accessible, an installing “wrapper” was written to make the pipeline easier for people unfamiliar with Nextflow.
Currently, the code has been tested only for Illumina paired-end and single-end data. The ViralFlow development team can not provide support for errors/problems of applying ViralFlow on sequencing reads from other platforms.
The publication of the first version of ViralFlow can be checked at this link. The publication of the version 1.0, which encompasses the functions described in this documentation, can be cheked here.
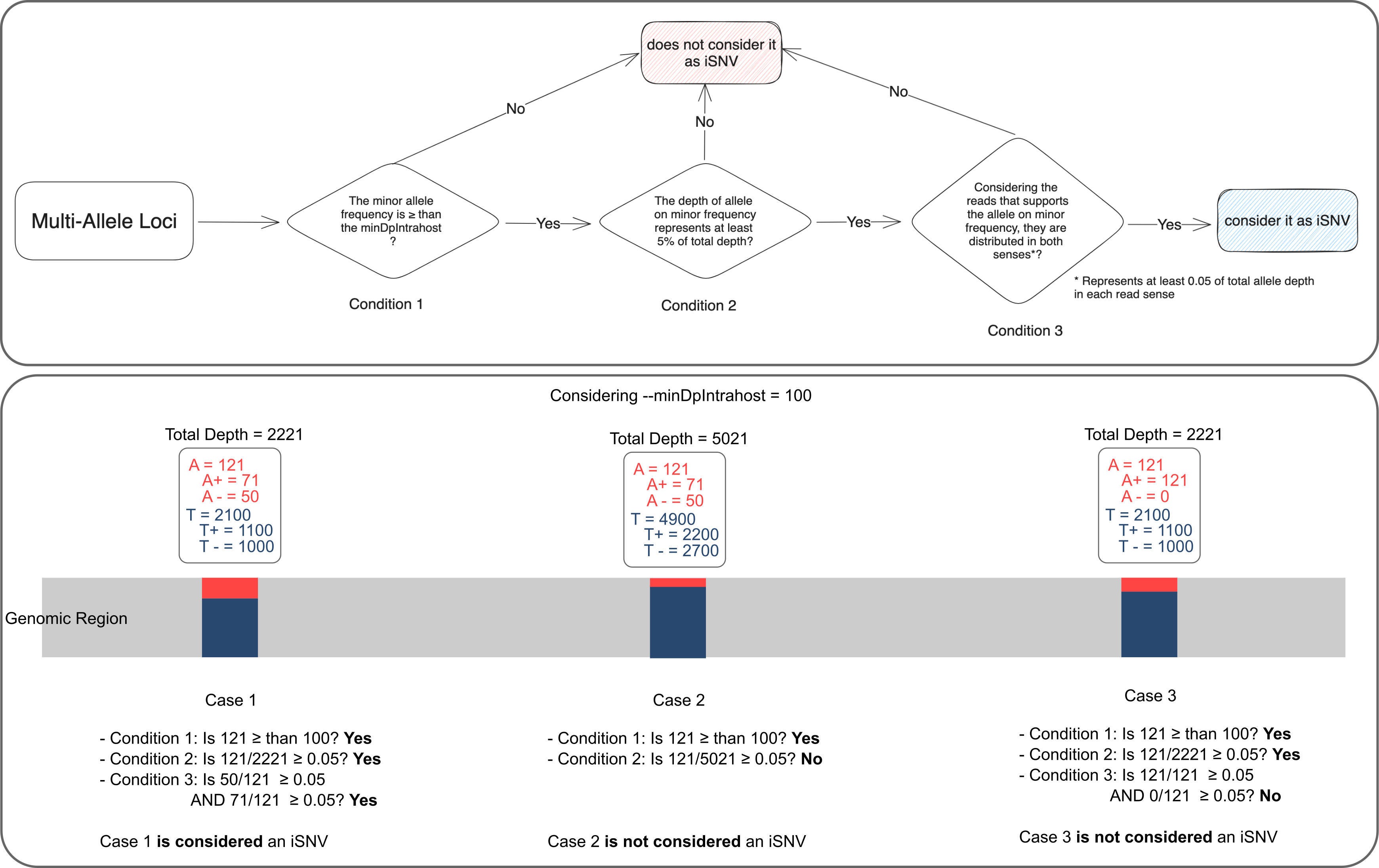
Intrahost Analysis
ViralFlow presents its own algorithm to detect iSNV (intrahost Single Nucleotide Variant) regions.
For a given multiallelic site to have a lower frequency allele considered as iSNV, three conditions must be met, these conditions are schematized in the upper part of the figure below.
n this logic, the bottom part of the figure below represents 3 multi-allelic sites and exemplifies the logic for considering which of them should be considered as iSNVs.
dependencies
To install and run ViralFlow, you will need at least 4.4Gb of disk storage (to build containers), and three dependencies:
- pip
- Git
- uidmap
The list with tool versions used in ViralFlow can be checked at this link. Moreover, for an installation with a controlled environment, we use Conda. ViralFlow uses the Singularity containerization tool and the Nextflow workflow language. To ensure that ViralFlow works correctly, the correct versions of Singularity and Nextflow are essential, both of which can be installed together with ViralFlow.
-
01 Conda
Conda is an open source package manager. It was initially developed to manage Python language package environments, allowing version control.
So that all users can use ViralFlow with the exact versions of Singularity and Nextflow, we created an environment with these versions, which allows a standardization in the ViralFlow installation environments, as well as the resolution of eventual errors.
-
02 Singularity v3.11.4
Singularity is a tool that performs virtualization at the operating system level, a task popularly known as containerization.
For a computational pipeline like ViralFlow to work, it requires several dependencies (other tools and libraries of the operating system) that can be complex to install. In order to make things easier to the user we use the container system which already comes with all dependencies needed to run ViralFlow. Therefore, the user does not need to install everything on his own.
With containerization, the user only needs to install one tool, in this case Singularity, and then build the containers based on recipe files (in the case of ViralFlow, this was also automated with the setupContainers.sh script). In this way, different research groups can run the tool with the same computational environment, without changes in ViralFlow's behavior, which guarantees the reproducibility of the results. -
03 Nextflow 22.04
Nextflow is a bioinformatics workflow manager that allows the development of portable and reproducible workflows.
Nextflow allows the execution of workflows in different computing environments, such as on-premises, high-performance computing (HPC) environments, cloud computing services, such as AWS and Google Cloud, and also in container orchestration systems, such as Kubernetes.
In addition, Nextflow provides support for managing dependencies with Conda, Docker, Podman or Singularity.
How to install (MacOS)?
Due to the limitation of using singularity on MacOS, to run ViralFlow on this type of system, we suggest using a Linux virtualization software called Lima. In this way, the user must follow three steps for installing Lima, and then satisfied with the Ubuntu installation guide.
Installing Lima
Install Lima using the Homebrew package manager:
brew install lima
Installing the Ubuntu instance
Follow the step by step installation of an Ubuntu instance by entering the command below
limactl start
Start Ubuntu
When the virtual machine is installed, use the command below to start Ubuntu and follow the ViralFlow installation steps as usual.
lima
How to install (Ubuntu)?
To install ViralFlow, four steps are necessary: Install system dependencies, in case you haven't installed them; Install Conda; install ViralFlow and, finally, assemble the containers for the analyses. This process is performed only once.
ViralFlow was developed and tested for the following operational systems and versions:
- Ubuntu 20.04 LTS;
- Ubuntu 22.04 LTS.
Installing system dependencies
If you don't have the pip dependency installer, the git version control system, and the uidmap package, you must install it with the following lines:
sudo apt update -y && \
sudo apt upgrade -y && \
sudo apt install curl git python3-pip uidmap -y
Installing and configuring the micromamba environment
We recommend managing micromamba environments due to their parallelization when downloading and installing dependencies. If you use another environment manager, such as conda, miniconda, mamba, etc., you can continue installing ViralFlow by skipping this step, however, conflicts during installation may occur due to versions of the specified dependencies, and their respective availability on different channels of resources.
If you don't have the micromamba environment manager installed, you can install and configure it with the following lines:
cd $HOME
curl -Ls https://micro.mamba.pm/api/micromamba/linux-64/1.5.7 | tar -xvj bin/micromamba
./bin/micromamba shell init -s bash -p ~/micromamba
source ~/.bashrc
micromamba activate
Installing ViralFlow
If you already have the aforementioned dependencies and conda installed, you can install ViralFlow with 5 lines of code:
git clone https://github.com/WallauBioinfo/ViralFlow
cd ViralFlow/
micromamba env create -f envs/env.yml
micromamba activate viralflow
pip install -e .
Building the containers.
All steps of ViralFlow are performed in controlled environments, where each tool will have the same version regardless of the research group that is using the tool. ViralFlow has its own method to carry out all this construction of environments, running just one line of code. For the building of containers, ViralFlow requires that the tool "unsquashfs" be available in the directory "/usr/local/bin/". During the container building process, a step to check for the presence of unsquashfs is performed. If the user's system does not have "unsquashfs" in the ideal location, ViralFlow will suggest the steps necessary to solve this problem. To avoid this, the user can create a symbolic link of the "unsquashfs" tool to the directory "/usr/local/bin/", for this the following command must be executed:
sudo ln -s /usr/bin/unsquashfs /usr/local/bin/unsquashfs
After ensuring that "unsquashfs" is in the appropriate location, run the command to build the containers:
viralflow -build_containers
How to run (Quick Guide)
ViralFlow provides 2 usage modes: sars-cov2 and custom. Regardless of the mode, the user must provide the absolute paths (the entire path to the directory or file to be indicated for the pipeline eg /home/user/test/) for each input file in the command line, otherwise the pipeline will be interrupted during execution. The explanation of each argument can be found here.
By default, the snpEff tool in ViralFlow is configured with the NC_045512.2 genome of the SARS-CoV-2 virus only. If you want to include the snpEff analysis for other viruses, you must update the snpEff database with the following line, example with Dengue:
viralflow -add_entry_to_snpeff --org_name Dengue --genome_code NC_001474.2
sars-cov2
In this model, the analysis is performed based on the reference genome NC_045512.2, and has, as additional analysis, the signature of strains with the Pangolin tool, and signing clades and mutations with the Nextclade tool. For this, the user needs to build a file with the analysis parameters, an example can be seen here.
viralflow -run --params_file test_files/sars-cov-2.params
custom
In this model, the analysis is performed based on the files for the virus that the user wants to analyze. In this mode, the user is responsible for providing each of the files necessary for the analysis. If the user wants to perform the snpEff analysis, he must pass the refseq code viral genome
viralflow -run --params_file test_files/denv.params
Pangolin update
Periodically the pangolin tool updates the lineage database, as well as the usher classification phylogeny, the scorpion mutation constellations, and the pangoLearn trained model. To update the tool and/or it's databases, just run ViralFlow with one of the commands:
#update the tool and databases
viralflow -update_pangolin
#update only the tool
viralflow -update_pangolin_data
Arguments
| Argument | Default value | Description |
|---|---|---|
| virus | sars-cov2 | Analysis type (sars-cov2 or custom). |
| primersBED | null | Absolute path to bed file with primers information used in genomic amplification (optional). |
| outDir | launchDir/output/ | Absolute path to the directory where the results will be stored. |
| inDir | launchDir/input/ | Absolute path to the directory with the input data (directory with the FASTQ files). |
| runSnpEff | true | Needed to run the snpEff tool (true or false). |
| writeMappedReads | true | Needed to generate the FASTQ files containing the sequencing reads that mapped to the reference genome. |
| minLen | 75 | Minimum size the reads must have. Reads below this thershould will be eliminated by FastP. |
| depth | 5 | Minimum coverage depth to call consensus bases. Positions with lower coverage depth will not be called and a “-” will be added to the respective consensus genomic position. |
| mapping_quality | 30 | Mapping quality treshold used to variant calling. |
| base_quality | 30 | Base quality treshold used to variant calling. |
| minDpIntrahost | 100 | Minimum coverage depth per genomic site to be considered in the intrahost analysis. |
| trimLen | 0 | Number of bases that should be trimmed at both ends of the reads. |
| refGenomeCode | null | Code of the genome to be used in the custom analysis. |
| referenceGFF | null | GFF genome file to be used in custom analysis. |
| referenceGenome | null | Fasta genome file to be used in custom analysis. |
| nextflowSimCalls | null | Number of simultaneous calls that nextflow can perform. |
| fastp_threads | 1 | Number of threads to be used in the fastp read filtering step. |
| bwa_threads | 1 | Number of threads to be used in the bwa mapping step. |
| mafft_threads | 1 | Number of threads to be used in the mafft alignment step. |
| dedup | false | This argument enable dedup mode of fastp. to activate it change value to true on params test file. |
| ndedup | 3 | When dedup mode is active you can use accuracy levels (1 - 6). You can change this value, but we recommend the standard. How much higher, more RAM and time are consumed. to activate it change value from 1 to 6 on params test file. |
Output files
The ViralFlow tool generates 2 types of output: specific and compiled.
Specific
For each sample in the analysis, a results directory will be created with the pattern `prefix_results` where the prefix is a code created from the fastq file name of the sample. Each directory has the following results:
| File | Description |
|---|---|
| prefix.all.fa.pango.out.csv | Tabular file with the results of the pangolin tool. |
| prefix.ann.vcf | file in tsv format (Tab separated value - comma-separated values) with annotation of the variants made by the snpEff tool. |
| prefix.depth5.all.fa.nextclade.csv | Tabular file with the results of the nextclade tool. |
| prefix.depth5.amb.fa | Consensus genome with ambiguous nucleotides at multiple allele positions. |
| prefix.depth5.fa | Consensus genome with majority nucleotides. Majority consensus, normally deposited in GISAID. |
| prefix.depth5.fa.algn | Consensus genome aligned with reference genome (following same size, mafft --keeplenght). |
| prefix.depth5.fa.algn.minor.fa | Consensus genome with minority nucleotides. |
| prefixo.depth5.fa.bc.intrahost.short.tsv | Tabular file summarizing the genomic positions where intrahost variants are supported. |
| prefix.depth5.fa.bc.intrahost.tsv | Tabular file with all information on intrahost variant positions. |
| prefix.mapped.R1.fq.gz | FASTQ R1 file with mapped reads. |
| prefix.mapped.R2.fq.gz | FASTQ R2 file with mapped reads. |
| prefix.metrics.genome.tsv | Tabular file with mapping depth and coverage metrics. |
| prefix.fastp.html | HTML file summarizing the results of the fastp tool. |
| prefix_snpEff_summary.html | HTML file summarizing the results of the snpEff tool. |
| prefix.sorted.bam | File with the sorted mapping of the sample reads against the reference genome. |
| prefix.tsv | vcf-like file generated by iVar. |
| prefix.unmapped.R1.bam.fq | FASTQ R1 file with unmapped reads. |
| prefix.unmapped.R2.bam.fq | FASTQ R2 file with unmapped reads. |
| prefix.vcf | vcf file. |
| metrics.alignment_summary_metrics | Text file with a summary of various metrics from the mapping. |
| nextclade.errors.csv | File reporting nextclade errors. |
| nextclade_gene_*.translation.fasta | File with the proteins of each gene. |
| snpEff_genes.txt | Tabular file with the number of variants and estimated impact per gene. |
| wgs | Textual file with mapping metrics, including depth by region. |
| prefixo_coveragePlot.png | PNG file with graphical visualization of genome coverage. |
| prefix_coveragePlot.svg | SVG file with graphical visualization of genome coverage. |
| prefix_coveragePlot.html | HTML file com a visualização gráfica da cobertura do genoma. |
| prefix_snpPlot.png | PNG file with a graphical visualization of the SNPs detected in the sample relative to the reference genome. |
| prefix_snpPlot.svg | SVG file with a graphical visualization of the SNPs detected in the sample relative to the reference genome. |
Compiled
At the end of the analysis, a directory named COMPILED_OUTPUT is generated with the compiled results, where:
| File | Description |
|---|---|
| errors_detected.csv | Samples with no resulting consensus. |
| lineage_summary.csv | It is generated for sars-cov- 2, presenting the lineage counts identified in the analysis, including lineages from minority genomes. |
| major_summary.csv | Tabular file with a summary of the depth, coverage and lineages (in case of sars-cov-2 analyses) of the majority consensus. |
| minor_summary.csv | Tabular file with a summary of the depth, coverage and lineages (in case of sars-cov-2 analyses) of minority consensus. |
| mutations.csv | Tabular file with the mutations found. |
| nextclade.csv | Tabular file with the result of nextclade (in case of sars-cov-2 scans). |
| pango.csv | Tabular file with the result of pangolin (in case of sars-cov-2 scans). |
| reads_count.csv | Tabular file with the count of reads per sample. |
| seqbatch.fa | Fasta file with majority consensus. |
| short_summary.csv | Tabular file with a summary of depth, coverage and lineage (in case of sars-cov-2 analyses) of both consensus.
|
| wgs.csv | Tabular file with picard output. |
| VF_REPORT/index.html | Summary HTML file with the compilation of fastp and snpEff results. |
Frequently Asked Questions
Should I mount containers for each analysis?
No, containers should only be rebuilt if there is an update of the workflow or specific tool of the pipeline (i.e. pangolin).
ViralFlow returned an error due to the absence of "unsquashfs" in the directory "/usr/local/bin/". What should I do?
A symbolic link for "unsquashfs" must be created. To do this, run the following command: sudo ln -s /usr/bin/unsquashfs /usr/local/bin/unsquashfs
Should I provide the adapters fasta file?
You can provide the fasta file with the PCR primers and/or adaptors in analyzes that use PCR amplification of viral genetic material prior to sequencing.
My ViralFlow review reported an error in the Pangolin update step, why?
This may happen if the Pangolin team releases a version with new dependencies, in which case you must rebuild the pangolin container with the line: singularity pull docker://staphb/pangolin
My installation went wrong in the step of mounting the containers, why?
What might have happened is that you have a different version of singularity than v3.11.4, you can check that with singularity --version. Another factor that can cause errors is that your internet network blocks query domains for building containers.
I installed the tool, my files are all organized following the manual, but the samples are not found, why?
ViralFlow recognizes samples by the suffixes _R1 or _R2 followed by .fq.gz or .fastq.gz, make sure your samples follow the required pattern. Examples:
- ART1_R1.fq.gz, ART1_R2.fq.gz will be recognized as the ART1 sample;
- ART1_denv_R1.fastq.gz, ART1_denv_R2.fastq.gz will be recognized as the ART1_denv sample.
Can I run ViralFlow on Windows??
Although some versions of Windows systems integrate a Linux interface through WSL (Windows Subsystem for Linux), our development team has not tested ViralFlow in these environments, therefore, the user is free to test the workflow in this type of environment, However, our team will not provide support for errors arising from these tests.
How to cite?
Currently, there are two publications on ViralFlow: the first, from 2022, covering the 'pre' 1.0 versions, and the second, from 2024, covering version 1.0.
Dezordi, F.Z.; Neto, A.M.d.S.; Campos, T.d.L.; Jeronimo, P.M.C.; Aksenen, C.F.; Almeida, S.P.; Wallau, G.L.; on behalf of the Fiocruz COVID-19 Genomic Surveillance Network. ViralFlow: A Versatile Automated Workflow for SARS-CoV-2 Genome Assembly, Lineage Assignment, Mutations and Intrahost Variant Detection. Viruses 2022, 14, 217. https://doi.org/10.3390/v14020217
Silva, A. F., Silva Neto, A. M., Aksenen, C. F., Jeronimo, P. M. C., Dezordi, F. Z., Almeida, S. P., Costa, H. M. P., Salvato, R. S., Campos, T. L., Wallau, G. L., on behalf of the Fiocruz Genomic Network. (2024). ViralFlow v1.0—a computational workflow for streamlining viral genomic surveillance. NAR Genomics and Bioinformatics, 6(2), lqae056. https://doi.org/10.1093/nargab/lqae056